Overview.
Post-excitation dynamics in biomimetic photo-catalysts. Strain-induced metal insulator transitions in strongly correlated transition metal oxide materials. Tracking ultrafast charge migration at a molecule-material interface with x-ray spectroscopy. Due in large part to the number of and complicated interactions between the electrons in systems like these, current theoretical methods are often unable to offer reliable predictions in support of the design and analysis of new experiments and technology. Our group specializes in developing new theoretical models, algorithms, and software in order to achieve reliable predictive power over ever larger and more complicated collections of electrons in molecules and materials.
Treating Excited States on an Equal Footing.
Due to both the historical priority of ground states and the inherent limitations of the traditional variational principle, electronic structure methods for modeling excited states tend to be more approximate than those for ground states. This imbalance is present both in chemistry, where excitations are most often modeled by linear response theory, and in solid state physics, where band gaps and spectra are most commonly derived from DFT and the many-body perturbation theory of Greens functions. While substantially more robust methods exist in both fields for modeling ground states (e.g. DMRG, QMC, fully relaxed coupled cluster, and so on), applying these methods to excited states is typically not possible without additional approximations, such as the linear response approximation or the use of state-averaged orbital shapes. A major direction of investigation in our group is into methods that exploit alternative variational principles that can treat excited states on the same footing as ground states, thus avoiding the need for different levels of approximation for different states. These approaches are especially important for excitations that frustrate traditional approaches, such as charge transfer excitations, band gaps near a Mott transition, and double excitations.
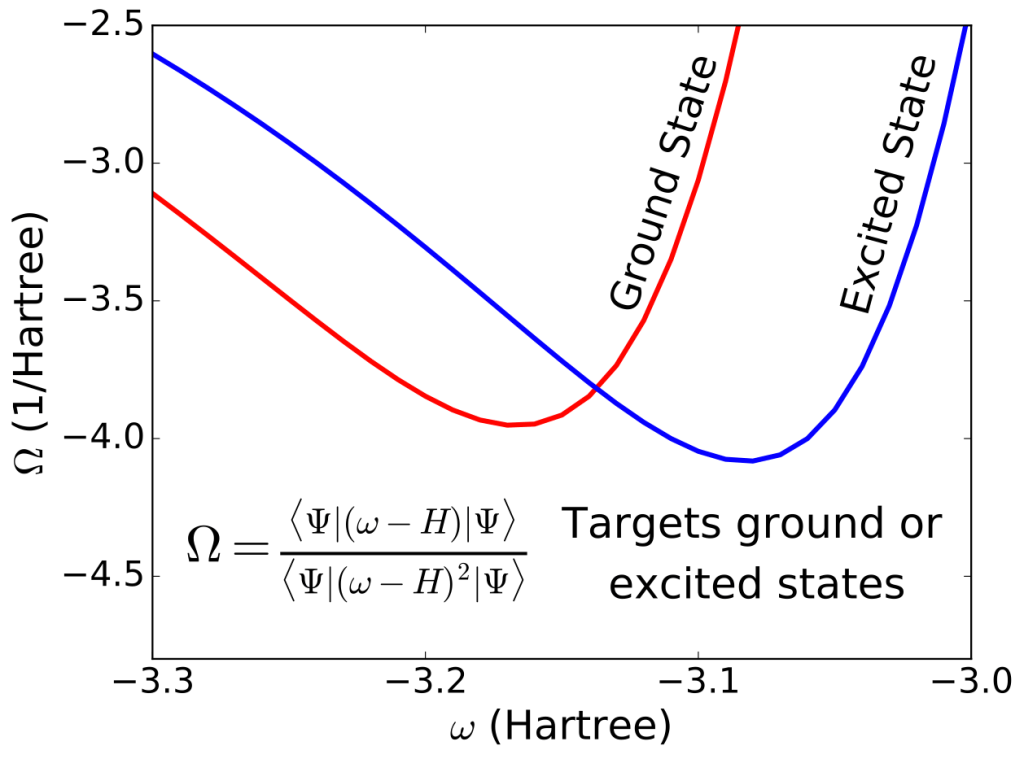
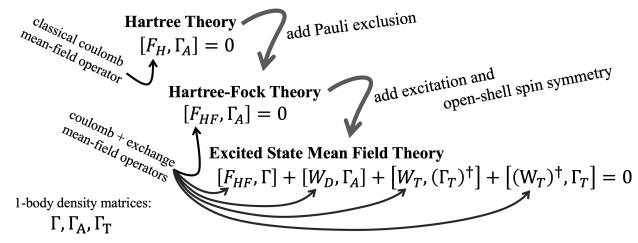
Excited State Mean Field Theory.
Post-excitation dynamics in biomimetic photo-catalysts. Strain-induced metal insulator transitions in strongly correlated transition metal oxide materials. Tracking ultrafast charge migration at a molecule-material interface with x-ray spectroscopy. Due in large ptarts from the Hartree product wave function and the classical coulomb interactions it encodes. As more quantum effects are added, the mean-field interactions become more complex, but a one-electron self-consistent field equation is maintained, even when we step firmly into the world of excited states. The obvious first step away from Hartree theory is to add Pauli exclusion, resulting in the non-classical exchange effects of Hartree-Fock theory. If we take the additional step of adding excited state character, we find that there are now four mean-field interactions instead of Hartree-Fock theory’s one, but the equations are still one-electron equations and can still be solved by efficient self-consistent techniques. This approach allows us to explore excited-state-specific effects like post-excitation orbital relaxations at mean-field cost. Further, this mean field theory can be used as a platform on which to build a wholly excited-state-specific hierarchy of correlation methods in a direct parallel to the role that Hartree-Fock theory plays in ground state quantum chemistry.
Developing New Wavefunction Approximations.
Whether dealing with ground or excited states, current methods for modeling large groups of electrons are either too expensive or too unreliable to provide the desired degree of predictive power in a wide range of molecular and materials systems. These include the multi-metal catalytic cores of many enzymes, various strongly correlated functional materials, and the charge-transfer relays responsible for moving electrons in and out of reaction centers in both natural and artificial photosynthesis. Our group is pursuing new directions in wave function approximation to help meet these challenges, including excited-state-specific perturbation theory, functional forms that can relax themselves in the presence of their own linear response, and wave function stenciling, in which one wave function component detects and deletes the unwanted portions of another. As in all areas of our work, this research requires careful consideration of the intersections between physical approximation and algorithmic efficacy.

Improving Underlying Algorithms.
Much of our group’s work is made possible by developing our own optimization algorithms so as to overcome the numerical challenges that arise from the physical approximations we employ. In some cases, we gain access to excited state variational principles though statistical sampling techniques whose random nature invalidates much of standard optimization theory. In other cases, we are faced with optimization problems that require high-order derivatives of energies and wave functions, which we have found can be evaluated efficiently for our purposes via recursive formulations of algorithmic differentiation. Regardless of the specific application, designing our algorithms to take proper advantage of modern supercomputers adds an additional layer of consideration and entertainment.
Quantum Monte Carlo.
We contribute to the QMCPACK software package, an open-source and highly-parallel suite of methods for quantum Monte Carlo simulations in molecules and materials. Two major goals of the wider QMCPACK community are to make QMC methods more black-box and to reduce their dependence on DFT-based molecular orbitals. We assist with both of these efforts by improving VMC optimization methods.
